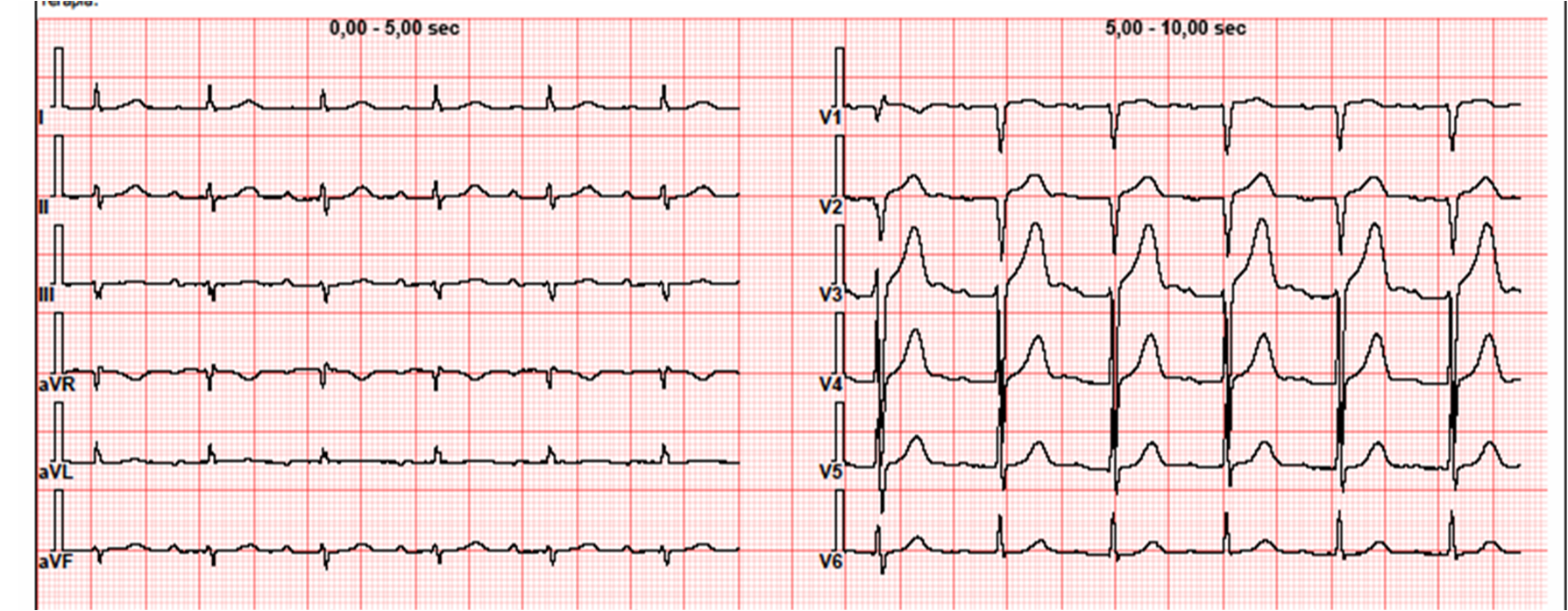
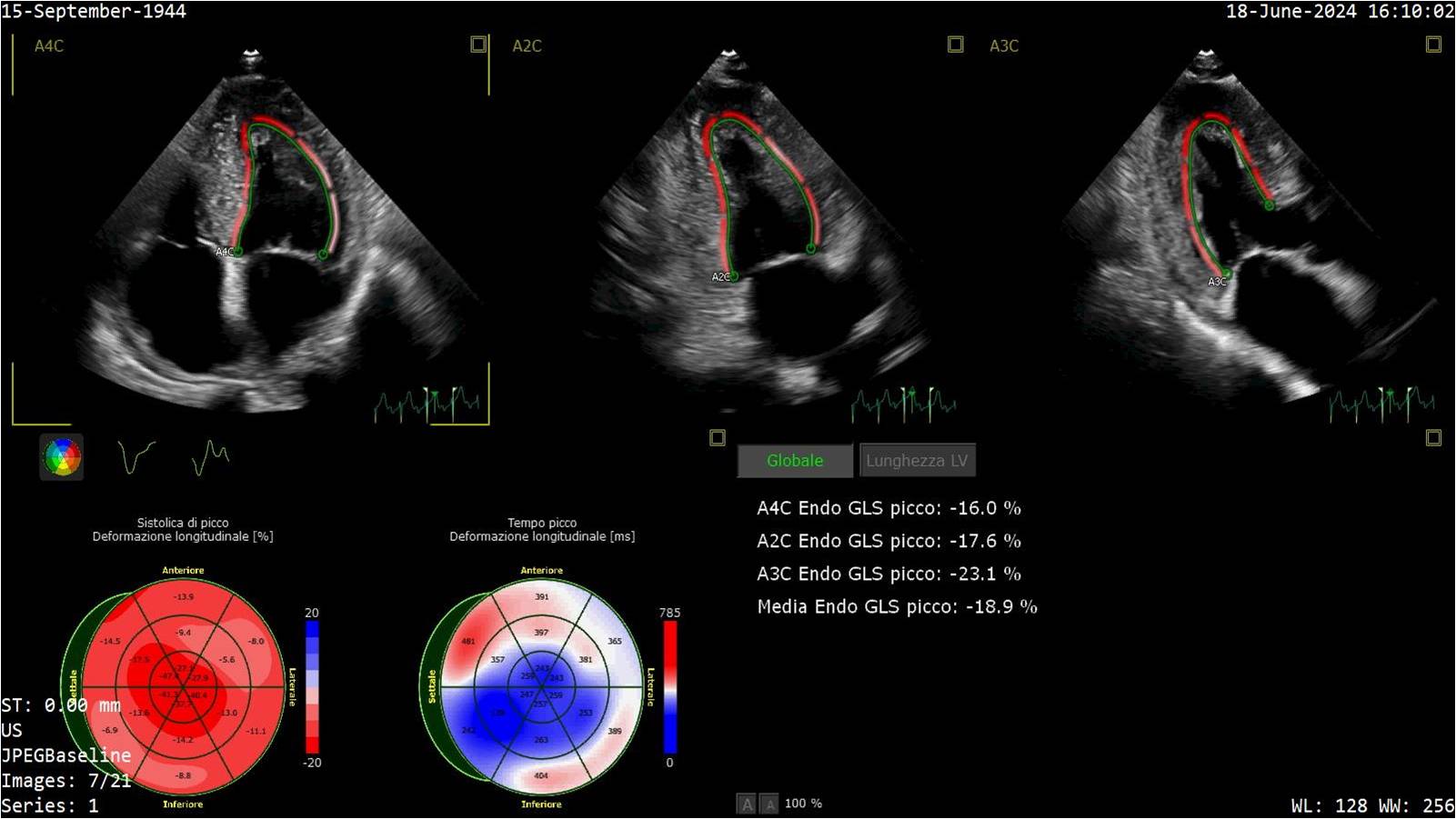
Introduction Hypertrophic cardiomyopathy (HCM) encompasses a spectrum of genetic cardiac disorders characterized by increased thickening of the myocardium. Amyloidosis, a buildup of amyloid, can coexist with or mimic hypertrophic cardiomyopathy, further complicating diagnosis and management. The study of HCM, including its intersection with amyloidosis, is crucial for understanding and treatment of these diseases (1). Clinical case In 2023 a 79-year-old male patient hypertensive and dyslipidemic underwent a cardiological evaluation for atrial arrhythmia. His medical history included lumbar canal stenosis requiring two surgeries and bilateral biceps tendon ruptures. ECG showed first-degree atrioventricular block and low voltages. Echocardiography revealed severe concentric hypertrophy (23 mm), normal ejection fraction, E/A ratio 0.8, increasing in E/e' ratio and PAPs 28+5 mmHg. Left ventricular strain was -18.6 with an apical sparing pattern, left atrial strain 20.6% and right ventricular strain -22.5%. The patient underwent a buccal swab for rapid TTR gene mutation testing which was negative. Blood tests showed NT-proBNP 990 pg/ml, Troponin HS 35 ng/L while serum and urinary immunofixation and light chains were normal. Bone scintigraphy was negative for myocardial uptake. Cardiac MRI revealed subendocardial fibrosis in all heart chambers and transmural fibrosis of the interventricular septum. Abdominal fat pad biopsy didn’t detect amyloid deposits. Endomyocardial biopsy evidenced large interstitial and perivascular fibrillar deposits, immunoreactive for anti-transthyretin polyclonal antibody. Genetic analysis showed a stopgain likely pathogenic variant in TTN gene. 24-hour Holter ECG revealed numerous atrial and ventricular extrasystoles. The patient covered 320 metres during 6MWT and scored 48 at Kansas City Cardiomyopathy Questionnaire. Cardiopulmonary exercise testing showed peak VO2 15.1 ml/kg/min (67% of predicted) and VE/VCO2 slope 29.47. Conclusions Given the histological diagnosis of cardiac amyloidosis, Tafamidis therapy was initiated. A careful follow-up was planned to monitor the response to therapy and assess the contribution of titin mutation and amyloid deposits to the patient's hypertrophy. Family screening was also recommended. References Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44 (37):3503-626.