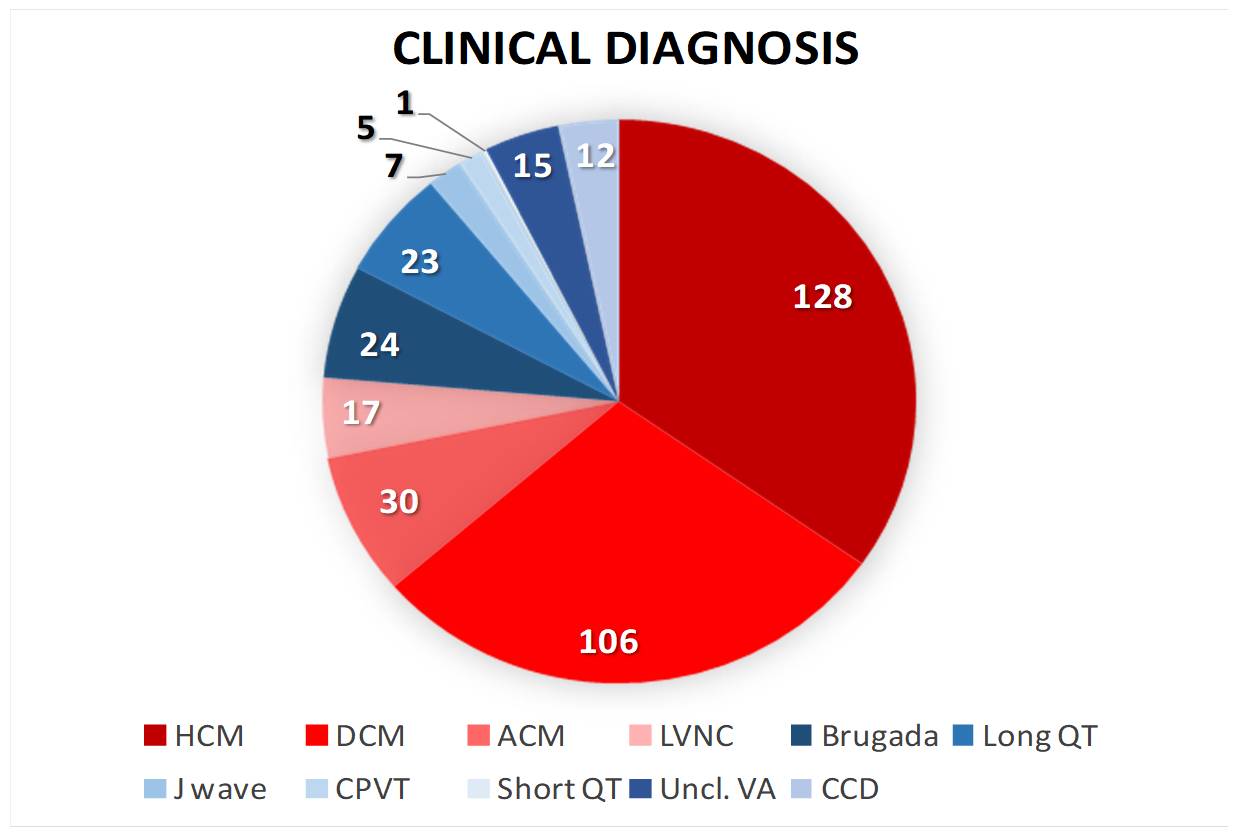
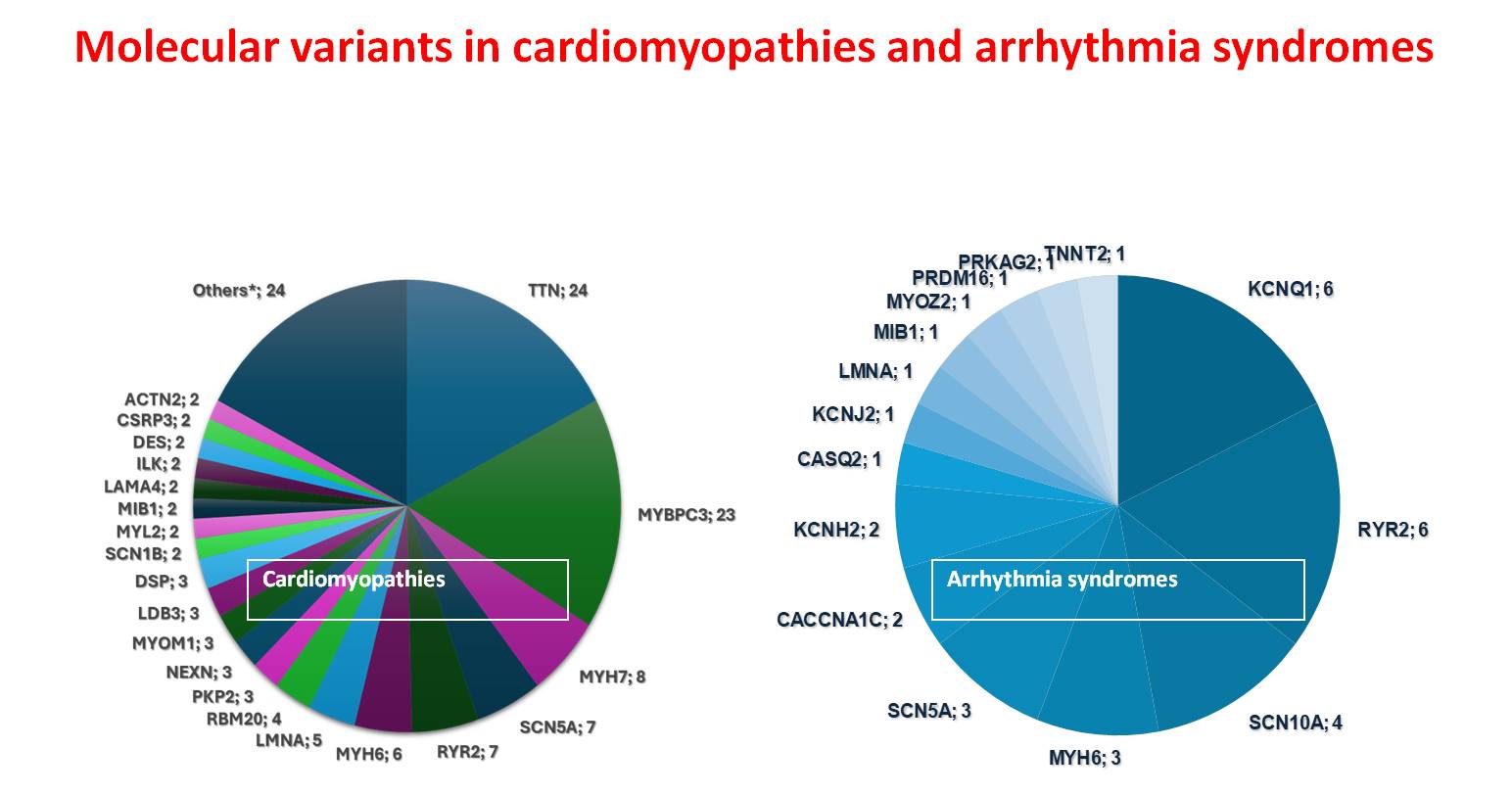
Inherited cardiac disorders encompass genetically diverse myocardial and electrical disturbances with substantial prognostic implications. This study investigated the genomic architecture and clinical manifestations of heritable cardiac conditions in a Southern Italian cohort of 363 families and 480 affected individuals. The phenotypic distribution showed predominance of structural myocardial disorders (69.95%) over cardiac ion channel dysfunction syndromes (30.05%). The clinical spectrum included hypertrophic cardiomyopathy (35%), dilated cardiomyopathy (29%), arrhythmogenic right ventricular cardiomyopathy (8%), ion channelopathies including long QT syndrome and Brugada syndrome (12%), and left ventricular non-compaction (5%), with minor phenotypes (<5%) comprising rare arrhythmogenic entities. Molecular characterization via targeted next-generation sequencing identified clinically relevant variants in 47.86% of cases, classified according to ACMG guidelines as pathogenic/likely pathogenic (P/LP) or variants of uncertain significance (VUS). The distribution of these genetic findings demonstrated a marked predominance in structural cardiac disorders (80%) compared to electrical cardiac disorders (20%). Despite extensive molecular analysis, 52.14% of the cohort remained genetically elusive, presenting no identifiable pathogenic variants associated with their cardiac phenotype. Sarcomeric and cytoskeletal protein alterations, specifically involving TTN and MYBPC3 loci, constituted one-third of structural cardiac pathologies, while ion channel disruptions in KCNQ1 and RYR2 accounted for one-third of electrical cardiac disorders. Integration of familial co-segregation studies, phenotypic characterization, and functional assessments enabled VUS reclassification and phenotype refinement. In other cases, genetic results allowed a different clinical and diagnostic framework. A notable case involved a 43-year-old proband with recurrent syncope, sustained polymorphic ventricular tachycardia, and post-immersion ventricular fibrillation. Initial diagnosis of CPVT was revised following identification of a pathogenic KCNJ2 variant. These findings underscore the importance of integrating comprehensive phenotyping with molecular diagnostics in managing inherited cardiac disorders, enabling improved diagnostic precision, therapeutic optimization, and refined risk stratification for sudden cardiac death.