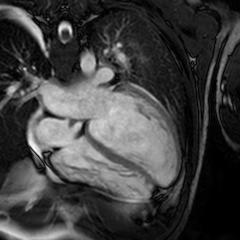
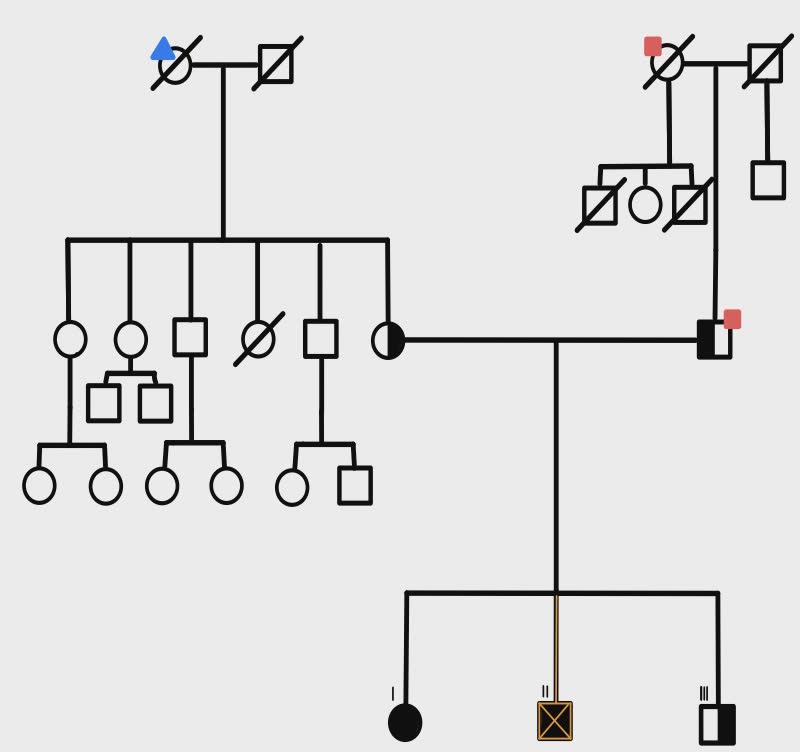
Intracellular Ca²⁺ regulation is critical for cardiac function, with SERCA2a pumping Ca²⁺ back into the sarcoplasmic reticulum for relaxation, counteracted by its reversible inhibitor phospholamban (PLN). Mutations in PLN, such as p.Leu39X, profoundly affect cardiac health. Homozygosity for Leu39X, described only twice in Greece (2001), is extremely rare and causes early-onset dilated cardiomyopathy (DCM) and heart failure, often requiring transplantation by the third decade. Heterozygotes display incomplete penetrance, with outcomes ranging from normal function to left ventricular hypertrophy or DCM. Case report. A 25-year-old male with no cardiovascular risk factors presented to the Emergency Department with progressive dyspnea and peripheral edema. Transthoracic echocardiography revealed severe biventricular dysfunction with significant mitral and tricuspid regurgitation. Comprehensive diagnostic workup excluded myocarditis, toxicological causes, and coronary artery disease. Cardiac magnetic resonance imaging (CMR) demonstrated late gadolinium enhancement (LGE) in the sub-epicardial inferolateral wall of the left ventricle, a finding subsequently confirmed by endomyocardial biopsy. Despite maximal medical therapy, the patient’s condition progressed to refractory cardiogenic shock, necessitating urgent heart transplantation. Post-transplant follow-up included genetic testing, which identified a homozygous variant NM_002667.5(PLN):c.116T>G (p.Leu39Ter) in the PLN gene. Genetic cascade screening of first-degree relatives revealed homozygosity for the same mutation in the patient’s older sister and heterozygosity in the younger brother and both parents, who are non-consanguineous. While all relatives were asymptomatic with normal echocardiograms and CMR findings (no evidence of LGE), 24-hour Holter monitoring detected non-sustained ventricular tachycardia in the asymptomatic sister. This case represents the third and fourth reported instances of homozygous p.Leu39Ter mutations in PLN-associated cardiomyopathy. It highlights the importance of genetic cascade screening in first-degree relatives of affected individuals to identify homozygous carriers at risk of developing the disease before clinical manifestation. Early detection enables close monitoring and proactive management strategies, which are critical in mitigating severe outcomes associated with phospholambanopathies.