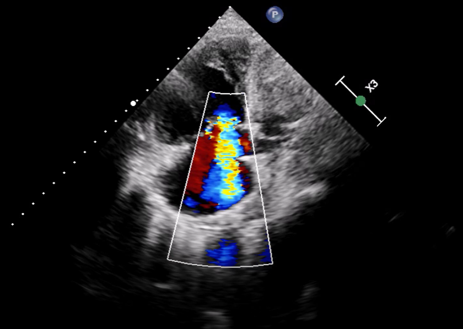
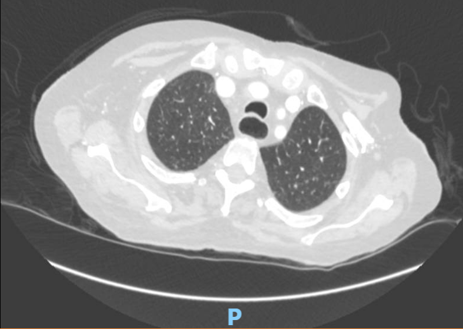
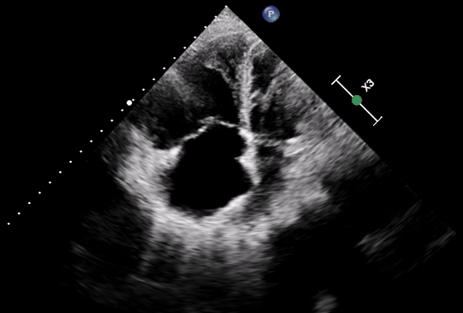
Pulmonary arterial hypertension (PAH) is a severe and life–threatening complication of systemic sclerosis (SSc), particularly challenging to diagnose in scleroderma sine scleroderma (ssSSc), a rare subset lacking the typical skin involvement. In ssSSc, early symptoms may be limited to Raynaud’s phenomenon, often overlooked, leading to delayed recognition of PAH and poor prognosis. SSc is an autoimmune disease characterized by vascular injury and fibrosis of the skin and internal organs, including lungs, heart, and kidneys. It is classified into diffuse cutaneous (dcSSc), limited cutaneous (lcSSc), and ssSSc, with ssSSc resembling lcSSc in autoantibody profile and visceral manifestations. We report a 60–year–old woman presenting with dyspnea and bilateral leg edema. Imaging revealed NSIP–type interstitial lung disease and dilated pulmonary arteries and heart chambers. NT–pro–BNP was elevated (8160 ng/ml), and ECG showed sinus rhythm with right axis deviation and right ventricular strain. Echocardiography demonstrated severe right heart failure with right ventricular dilation (40 mm), interventricular septum flattening (EI 1.5), severe tricuspid regurgitation, and markedly impaired RV function (FAC 21%, TAPSE 13 mm, TAPSE/PAPs 0.14 mm/mmHg). Her history included diabetes, repeated COPD–like exacerbations, Raynaud’s phenomenon, and mild dysphagia. Reevaluation of CT imaging revealed subtle esophageal dilatation, prompting autoimmune testing, which showed high–titer ANA (1:2560) with nucleolar pattern. Based on clinical, imaging, and serological findings, ssSSc was diagnosed. Right heart catheterization confirmed significant precapillary pulmonary hypertension. Despite aggressive therapy, including non–invasive ventilation, parenteral prostanoids, and inhaled nitric oxide, the patient progressed to respiratory failure and died. This case underscores the importance of considering ssSSc in patients with SSc–like visceral involvement even in the absence of cutaneous manifestations. Lack of skin changes contributes to delayed diagnosis, allowing irreversible vascular and organ damage. Early recognition and timely initiation of therapy are critical to enhance survival. Awareness among clinicians—including cardiologists, pulmonologists, and rheumatologists—is essential to ensure prompt multidisciplinary management and optimize prognosis and quality of life in patients with this rare and severe form of systemic sclerosis.