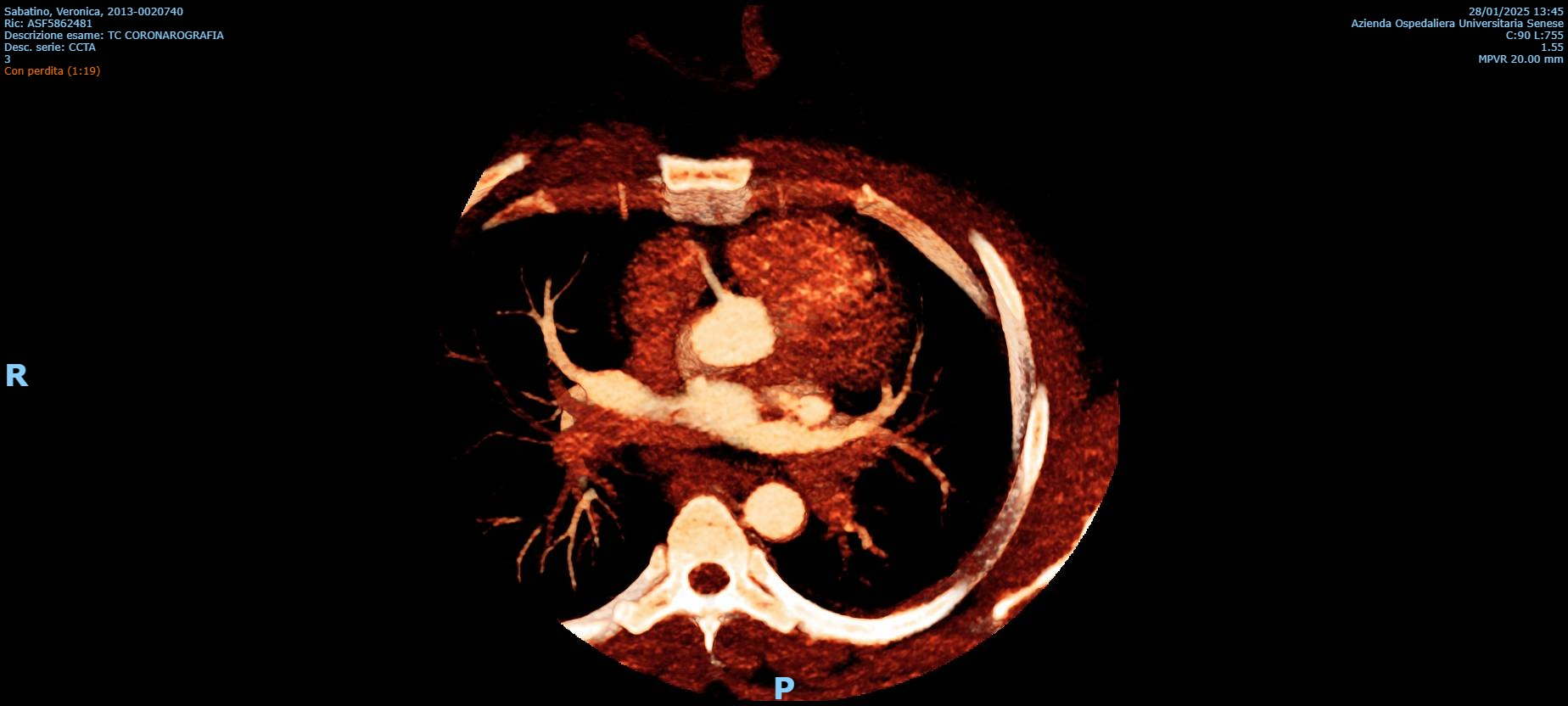
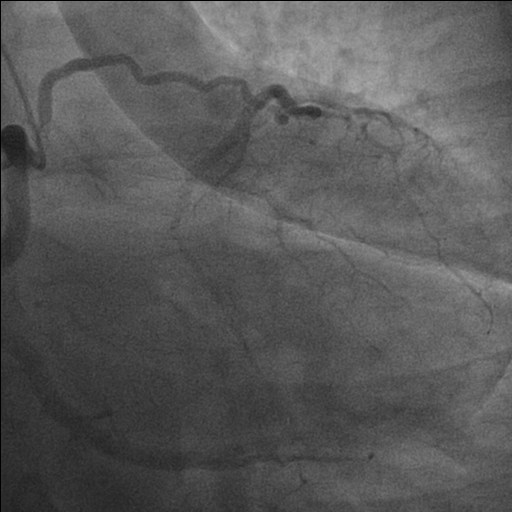
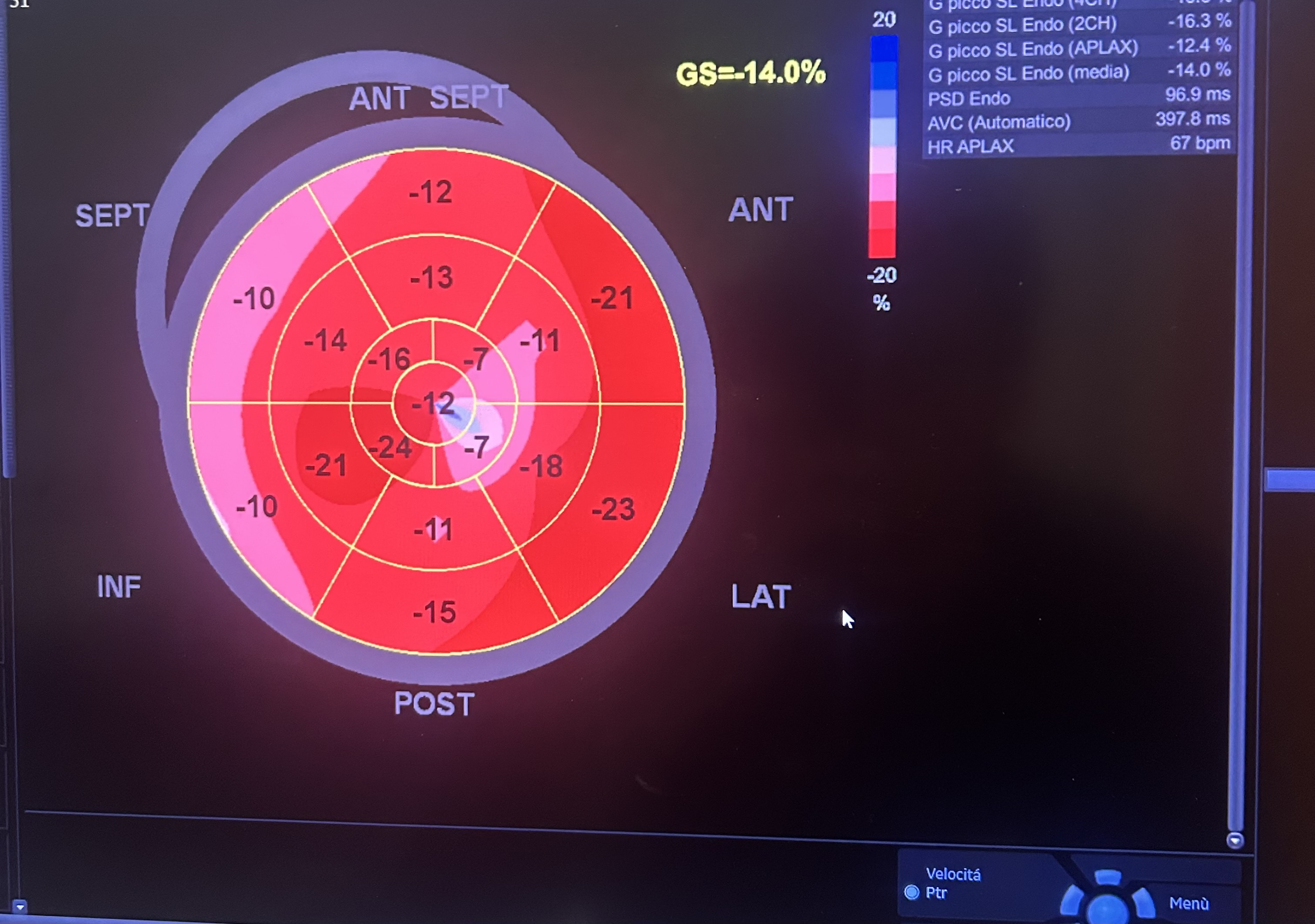
Background: Congenital absence (agenesis) of the left main coronary artery (LMCA) is an exceptionally rare coronary anomaly. Its coexistence with dilated cardiomyopathy (DCM) is scarcely reported, and the pathophysiological relationship between these conditions remains uncertain. Case presentation: A hypertensive female patient with multiple cardiovascular risk factors presented with exertional angina, palpitations, dyspnea on moderate exertion, and asthenia. Transthoracic echocardiography revealed mild left ventricular (LV) dilation with borderline systolic function and reduced global longitudinal strain, consistent with hypertensive heart disease. Coronary computed tomography angiography showed congenital absence of the left coronary artery, with compensatory collateral circulation originating from the conus branch supplying the anterior LV wall. Invasive coronary angiography confirmed the absence of the LMCA, a dominant right coronary artery, and collateral flow to a hypoplastic left anterior descending artery and the circumflex artery. Cardiac magnetic resonance imaging demonstrated preserved biventricular volumes and function, mild regional wall motion abnormalities, and subepicardial delayed gadolinium enhancement with a non-ischemic pattern in the mid-basal lateral wall. At follow-up, echocardiography showed progression to mildly reduced LV systolic function (LVEF 45%), consistent with ischemic–hypertensive cardiomyopathy. The patient’s twin sister, affected by dilated cardiomyopathy, underwent coronary CT angiography, which demonstrated anatomically normal coronary arteries. Given the familial occurrence of DCM, genetic testing was performed in both sisters to investigate a possible hereditary etiology. Discussion and conclusions: This case highlights the diagnostic complexity of concomitant LMCA agenesis and DCM. It remains unclear whether these conditions represent two independent entities sharing a genetic background or whether chronic myocardial hypoperfusion related to anomalous coronary anatomy may have contributed to LV remodeling. The presence of non-ischemic subepicardial fibrosis suggests additional mechanisms beyond epicardial coronary supply. Multimodality imaging and genetic evaluation are essential to clarify the relationship between congenital coronary anomalies, myocardial fibrosis, and ventricular dysfunction.