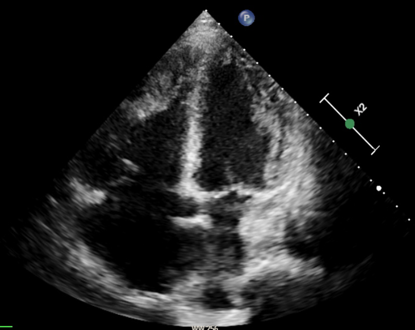
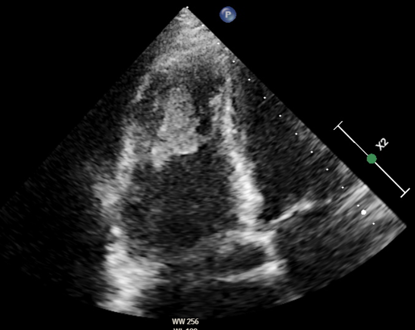
Autoimmunity plays a central role in the pathogenesis of idiopathic inflammatory myopathy (IIM), with autoantibodies identified in over 50% of patients. These target both nuclear and cytoplasmic cellular components and are classified as myositis–specific (MSAs) or myositis–associated autoantibodies (MAAs), often present in other connective tissue diseases (CTDs). While pulmonary hypertension (PH) is a recognized complication of CTDs, it is rare in IIM, and when present, it is frequently associated with interstitial lung disease. The prevalence of MSAs/MAAs in PH patients remains unclear. We report a 70–year–old woman admitted for dyspnea, ascites, jugular vein distension, and leg edema. She had a two–year history of dysphagia, weight loss, and mild myalgia. Laboratory evaluation showed markedly elevated NT–pro–BNP and ECG revealed atrial fibrillation with right ventricular strain. Chest CT demonstrated nonspecific ground–glass opacities and bilateral distal pulmonary emboli. Echocardiography showed severe right heart dilation and dysfunction, consistent with right heart failure due to precapillary PH, with evidence of right intraventricular thombosis. Extensive laboratory testing revealed positivity for lupus anticoagulant (LAC), prompting anticoagulation with warfarin. Notably, antinuclear antibodies (ANA) were negative, whereas anticytoplasmic autoantibodies (AC–15, AC–16, AC–18) were detected. The interplay between myopathy and PH is multifactorial. Skeletal, particularly respiratory, muscle impairment can induce hypoventilation and hypoxemia, promoting PH onset or progression. Inflammatory and autoimmune mechanisms may directly affect pulmonary vasculature, causing endothelial injury and remodeling. Esophageal involvement can produce dysphagia. Diagnosis remains challenging due to clinical heterogeneity and overlap with other neuromuscular or systemic diseases. Cytoplasmic autoantibodies, though less commonly reported than nuclear patterns, may provide critical diagnostic information. This case illustrates the complexity of PH in CTDs, where overlapping pathophysiological mechanisms may place patients along a spectrum between Group 1 and Group 4 chronic thromboembolic PH. It underscores the importance of extending immunological workup beyond conventional autoantibodies, as less frequently investigated anticytoplasmic antibodies can reveal rare forms of PH associated with mixed connective tissue disease or inflammatory myopathies.