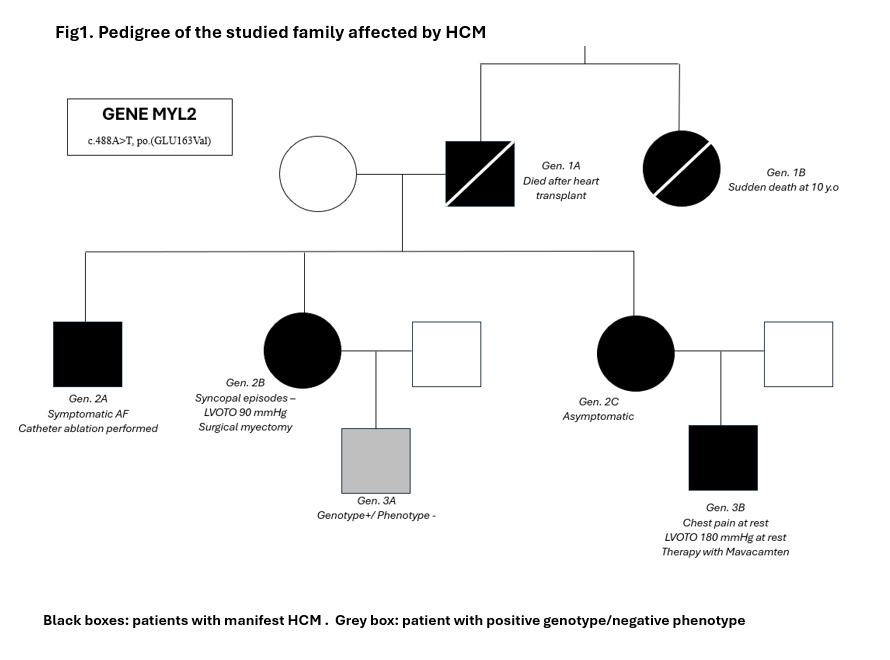
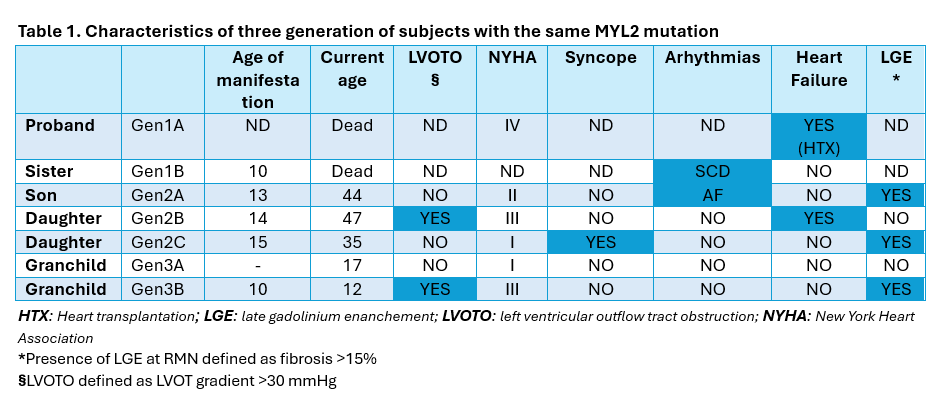
Introduction Hypertrophic cardiomyopathy (HCM) is a cardiac disease characterized by increased ventricular wall thickness in the absence of abnormal loading conditions. Most cases are associated with genetic variants encoding sarcomeric proteins. 964 variants have been identified in over 25 HCM-associated genes, most frequently involving MYH7 and MYBPC3. Variants in the MYL2 gene, encoding the myosin regulatory light chain, are rare (1-2%). Among these, some missense variants are classified as pathogenic, while others remain unclassified due to the variable penetrance in HCM expression. Case description We identified a family carrying a mutation for MYL2 gene (not described in the Literature, but defined “likely pathogenetic”) to show how the same mutation can determine a heterogeneous spectrum of HCM-related clinical manifestations within the same family nucleus. The father, proband, (Fig1.Gen1A) affected by HCM with a phenotype that evolved into a dilated form, underwent heart transplantation in 1998 and died on day 21 from post-surgical complications, while his sister (Fig1.Gen1B) died suddenly at age 10. The three children (Fig1.Gen2) manifested HCM around age 16 (IVS > 20 mm), but with different phenotypes: a son (Fig1.Gen2A) presented, at age 28, a non-obstructive arrhythmogenic form with over 5 episodes of atrial fibrillation, treated with electrical cardioversion and subsequently with catheter ablation; a daughter (Fig1.Gen2B) at age 35 developed a severe obstructive form with recurrent syncopal episodes requiring myectomy and mitral plasty; another daughter (Fig1.Gen2C) remains asymptomatic without therapy. Of the two grandchildren (Fig1.Gen3), one (Fig1.Gen3A) is genotype-positive but phenotype-negative at age of 17, while the other one (Fig1.Gen3B) at 10 years showed rapid ventricular thickening (> 10 mm in one year) with development of obstructive phenotype (Gmax at rest > 150 mmHg) with chest pain for which Mavacamten therapy at nominal use was started. Conclusions This family case highlights that a single MYL2 mutation can result in marked phenotypic variability even within the same family. It raises several questions: Can the environmental factors play a role in the development of a given phenotype? Can there be mutations at other gene loci that played a pejorative role in the manifestation of the pathology? In conclusion, HCM requires a personalized clinical approach and specific follow-up, even when the mutation is shared.