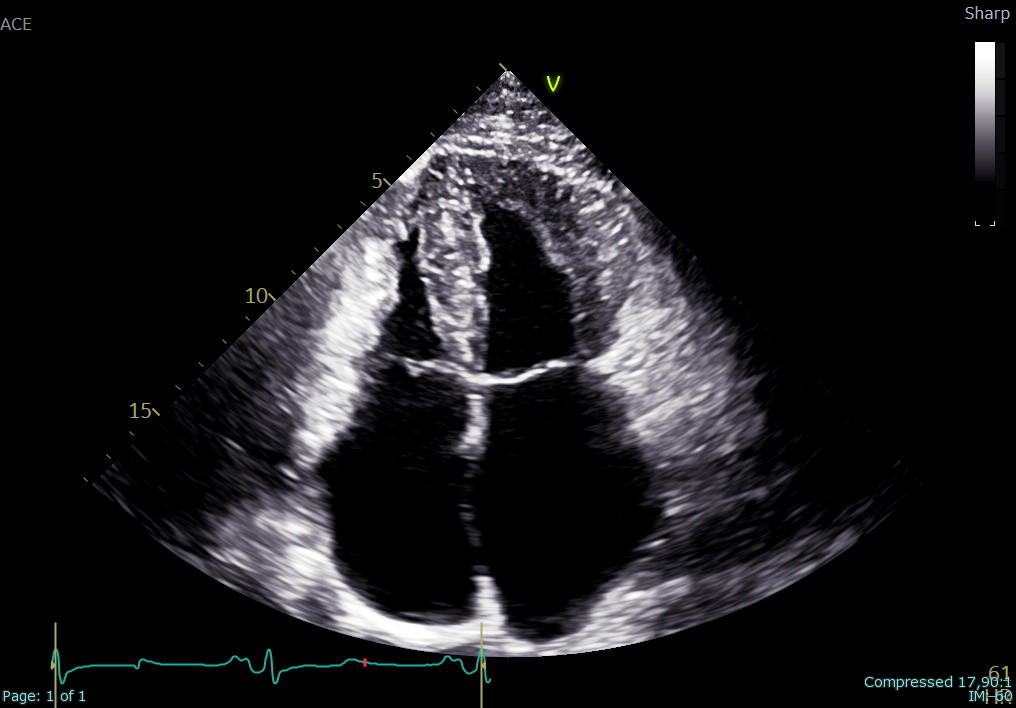
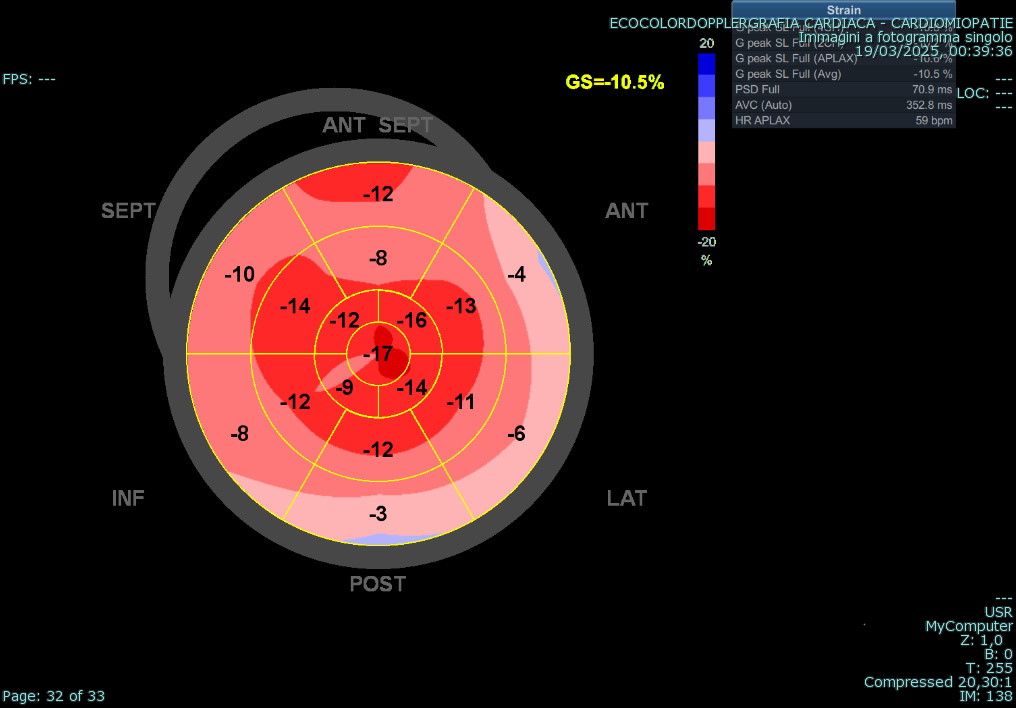
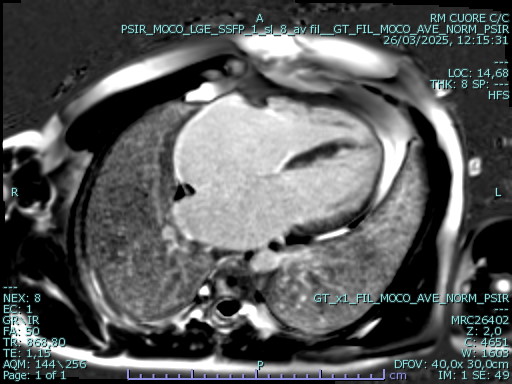
An adult patient was referred for cardiological assessment due to a complex family history of cardiac disease. Two of his five children, who had structural cardiac abnormalities, were being followed for suspected long QT syndrome despite negative genetic testing and one of his cousins died suddenly at 44 years of age. In the index patient, baseline ECG showed diffuse repolarization abnormalities with QTc prolongation. Laboratory testing revealed markedly elevated levels of NT-proBNP, while 24-hour Holter monitoring showed no ventricular arrhythmias. Transthoracic echocardiography (file 1) demonstrated small ventricular cavities, severe diastolic dysfunction with elevated filling pressures, mildly reduced systolic function, reduced longitudinal strain with an apical sparing pattern (file 2), and severe bi-atrial dilation. Trabecular myocardial remodeling resulted in an apparent concentric increase in left ventricular wall thickness, consistent with pseudo-hypertrophy rather than true hypertrophic remodeling. Cardiac magnetic resonance (file 3) revealed marked biventricular apical trabeculation fulfilling criteria for non-compaction, mildly reduced global biventricular systolic function, severe biatrial enlargement, and areas of non-ischemic myocardial late gadolinium enhancement. Associated pericardial thickening with late enhancement was also observed. Right heart catheterization confirmed isolated post-capillary pulmonary hypertension at rest and during exercise, secondary to left ventricular diastolic dysfunction. Further investigations for infiltrative cardiomyopathies, including iron panel, serum and urinary immunofixation, and free light chain assessment, were negative. Genetic testing using an extended cardiomyopathy panel, including TTR and GLA, was negative. Subsequent analysis by array-CGH identified a duplication in chromosome 7q22.3 involving the KMT2E gene, associated with a rare autosomal dominant neurodevelopmental disorder. This case demonstrates how a restrictive pattern may obscure the underlying complexity of myocardial disease characterized by trabecular remodeling, pseudo-hypertrophy, and a possible syndromic genetic background. A comprehensive diagnostic strategy is essential to ensure accurate characterization and to guide tailored patient management.