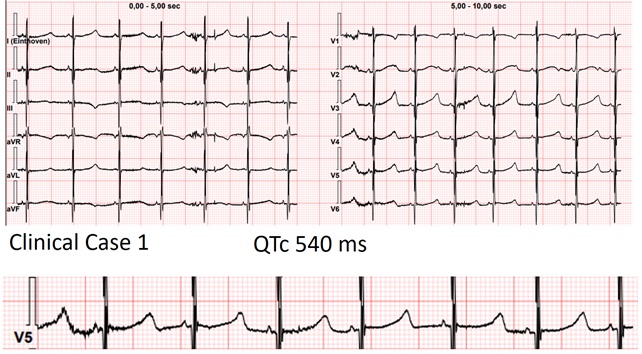
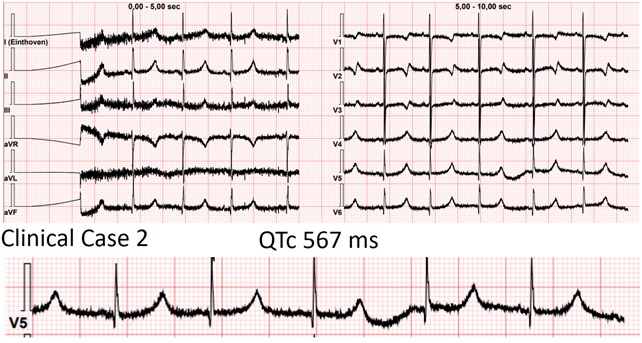
Background Jervell and Lange-Nielsen syndrome (JLNS) is the autosomal recessive form of long QT syndrome characterized by congenital deafness and a high risk of malignant arrhythmias in early childhood. We report 2 cases of preschool children highlighting clinical presentation and differences in arrhythmic triggers. Case 1 A 2-year-old boy, followed since birth for JLNS. Parents were first-degree cousins with a tragic family history of long QT syndrome and sudden cardiac death (SCD. Serial evaluations consistently showed a markedly prolonged QTc, poorly responsive to maximal propranolol therapy, despite no major arrhythmias. During follow-up, he experienced a syncopal episode in the context of severe iron-deficiency anemia. ECG at that time showed QTc ~640 ms, which return to his normal levels (~520 ms) after red blood cell transfusion. No major arrhythmias were documented, but the event was considered facilitated by anemia on a high-risk substrate. Subsequent follow-up showed clinical stability. Case 2 A 3-year-old girl, followed since infancy for JLNS. The parents were first-degree cousins, both affected by long QT syndrome. Serial ECGs showed persistently prolonged QTc (~530 ms) without major arrhythmias. She was treated with maximal-dose nadolol. During follow-up, she experienced syncope associated with severe hypoglycemia, likely induced by beta-blocker therapy. Inadequate nutrition, including frequent omission of breakfast, was identified. The event resolved after dietary counseling with subsequent stability. Discussion These cases reflect the high arrhythmic risk in JLNS, consistent with literature reporting life-threatening events in ~30% of children within the first 6 years. Despite identifiable and potentially modifiable triggers, the intrinsic arrhythmic risk led to consideration of implantable loop recorder. In both cases, small body size precluded implantation, highlighting a critical limitation in device availability. Conclusions Key similarities include parental consanguinity, family history of SCD, persistently prolonged QTc despite maximal beta-blocker therapy, and the need for close monitoring. Differences lie in the triggers of syncope: severe anemia and beta-blocker–induced hypoglycemia. These cases underscore the importance of a multidisciplinary approach, combining optimized pharmacologic therapy, family education, continuous monitoring, and management of extracardiac factors to reduce the risk of lethal arrhythmic events.