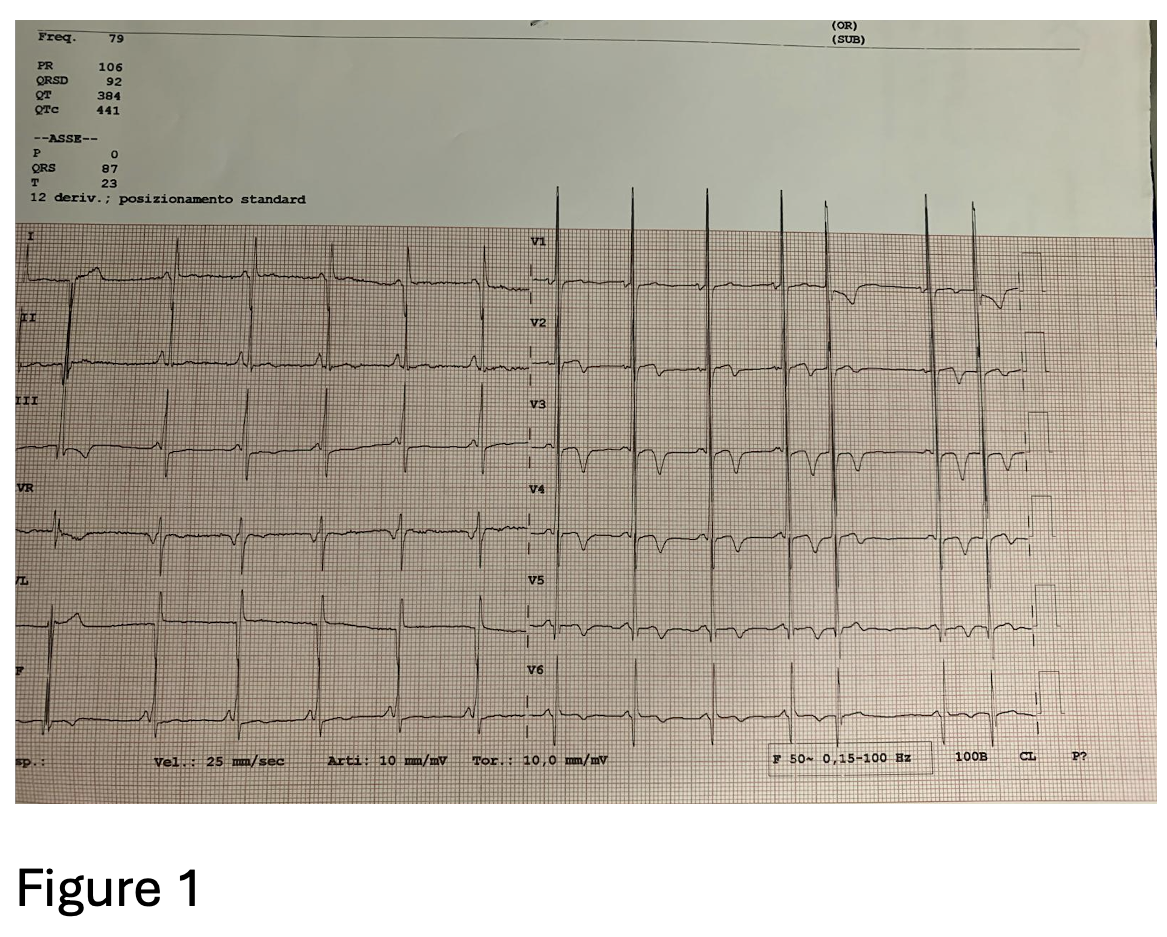
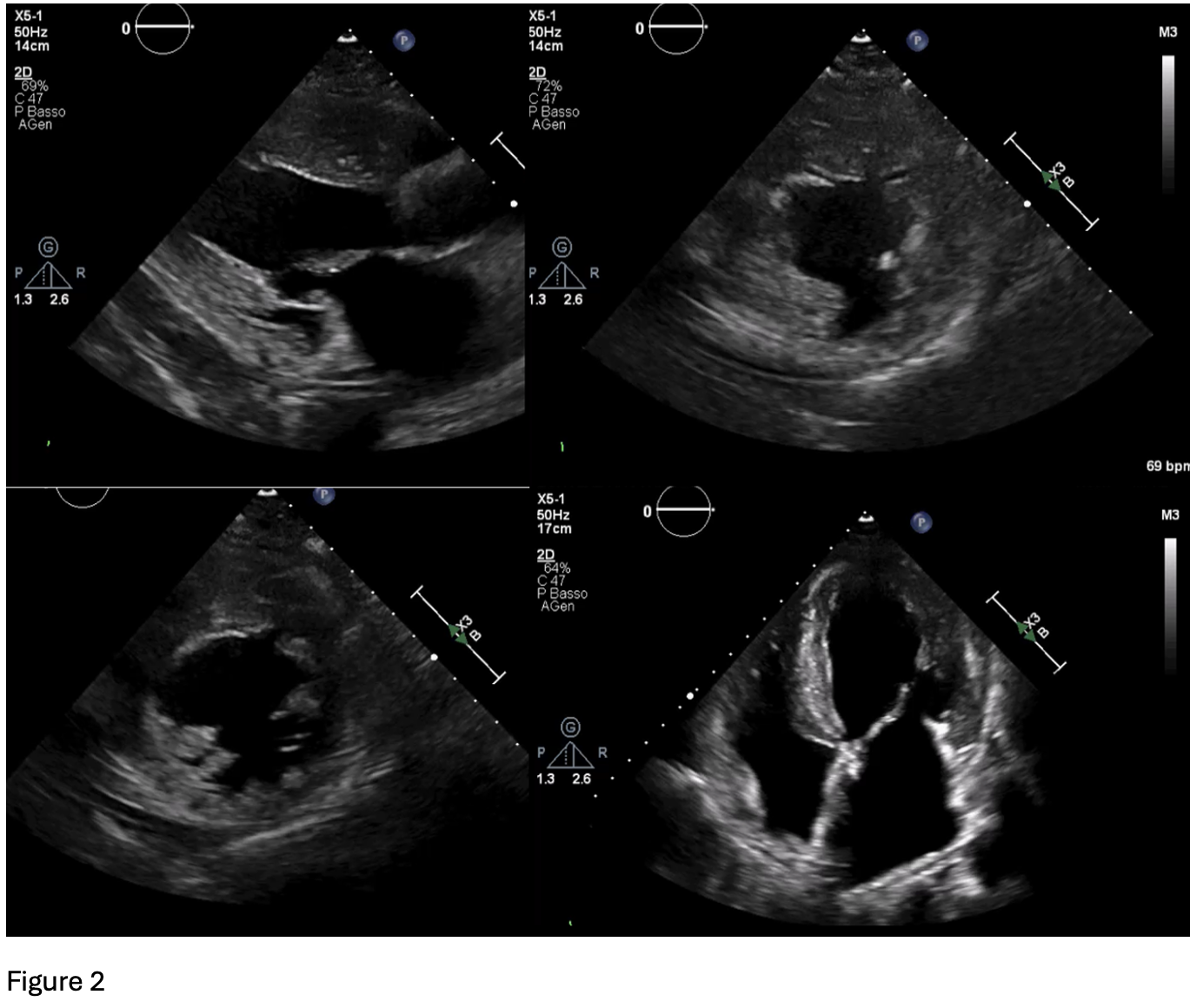
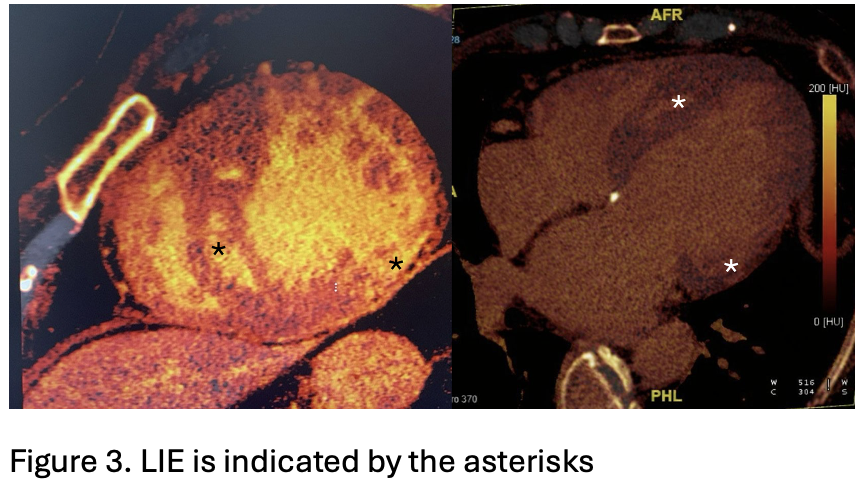
A 64-year-old woman with maternally inherited hypertrophic cardiomyopathy (MIHCM) was referred to our Cardiomyopathy Center. She was diagnosed with HCM at age 30 via family screening, initially presenting with moderate hypertrophy (IVS 16 mm, PW 13 mm) and preserved left ventricular (LV) systolic function. Family history was significant for adverse cardiac events: her mother died at age 54 from heart failure (HF) in the hypokinetic phase, and two male cousins required heart transplantation at a young age. Genetic testing previously identified a mtDNA A4300G mutation in the tRNAIle gene, which is specifically associated with MIHCM and isolated cardiac involvement. Clinical Course The patient’s follow-up was irregular until January 2025, when she was hospitalized for pulmonary edema. Following stabilization, she presented to our center with NYHA Class II-III dyspnea. Medications included metoprolol, furosemide and canrenone. ECG showed sinus rhythm with a short PR interval, deep Q-waves (DI, aVL, V6), diffuse T-wave inversion, left atrial enlargement, and marked biventricular hypertrophy (Fig.1). Echocardiography revealed concentric hypertrophy—maximal at the mid-anterior septum (24 mm) and posterior wall (13 mm)—with a reduced LVEF of 38%, diffuse hypokinesia, and a GLS of -9%. Diastolic dysfunction was severe (E/e’ 27) with a dilated left atrium (55 ml/m²) (Fig.2). Laboratory results showed elevated NT-proBNP (3114 pg/ml) and hs-TnI (103 ng/L). Angiography excluded coronary artery disease; right heart catheterization confirmed hemodynamic congestion and reduced cardiac index (PCWP 20 mmHg; mPAP 26 mmHg; CI 2.2 L/min/m²). Due to claustrophobia, cardiac CT with Late Iodine Enhancement (LIE) was performed instead of MRI, revealing extensive intramyocardial fibrosis in the mid-apical septum and inferolateral wall (Fig.3). Management and Conclusions An ICD was implanted for primary prevention. HF therapy was optimized with the addition of an SGLT2 inhibitor and low-dose ARB, though up-titration was limited by symptomatic hypotension. The patient was subsequently referred to the Advanced Heart Failure clinic. This case highlights a specific phenotype of mtDNA-related MIHCM: matrilinear transmission, non-obstructive concentric hypertrophy (often involving the posterior wall) and a high risk of progression to LV dysfunction. Clinicians should consider mitochondrial mutations in patients with "sarcomere-negative" HCM, even in the absence of multisystemic symptoms.